Reaction mechanisms and catalysis
Chemical Reaction Engineering
Heterogeneous catalysis at different scales
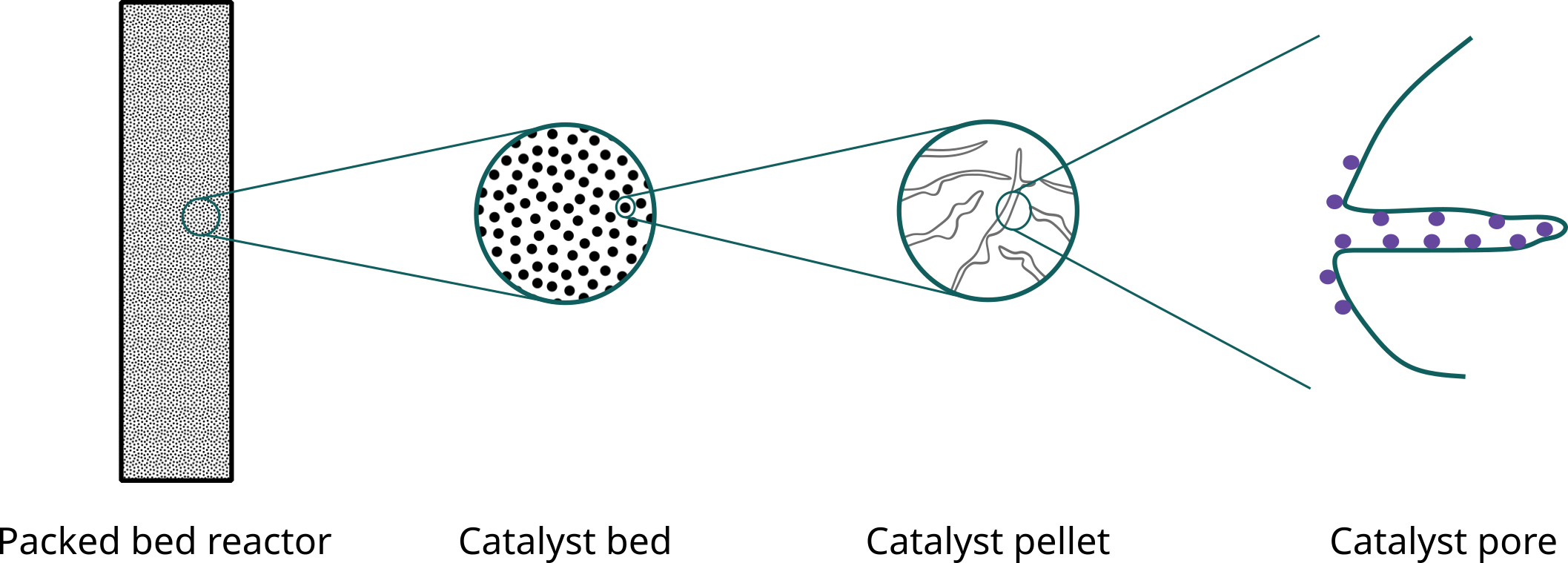
Steps in a heterogeneous catalytic reaction
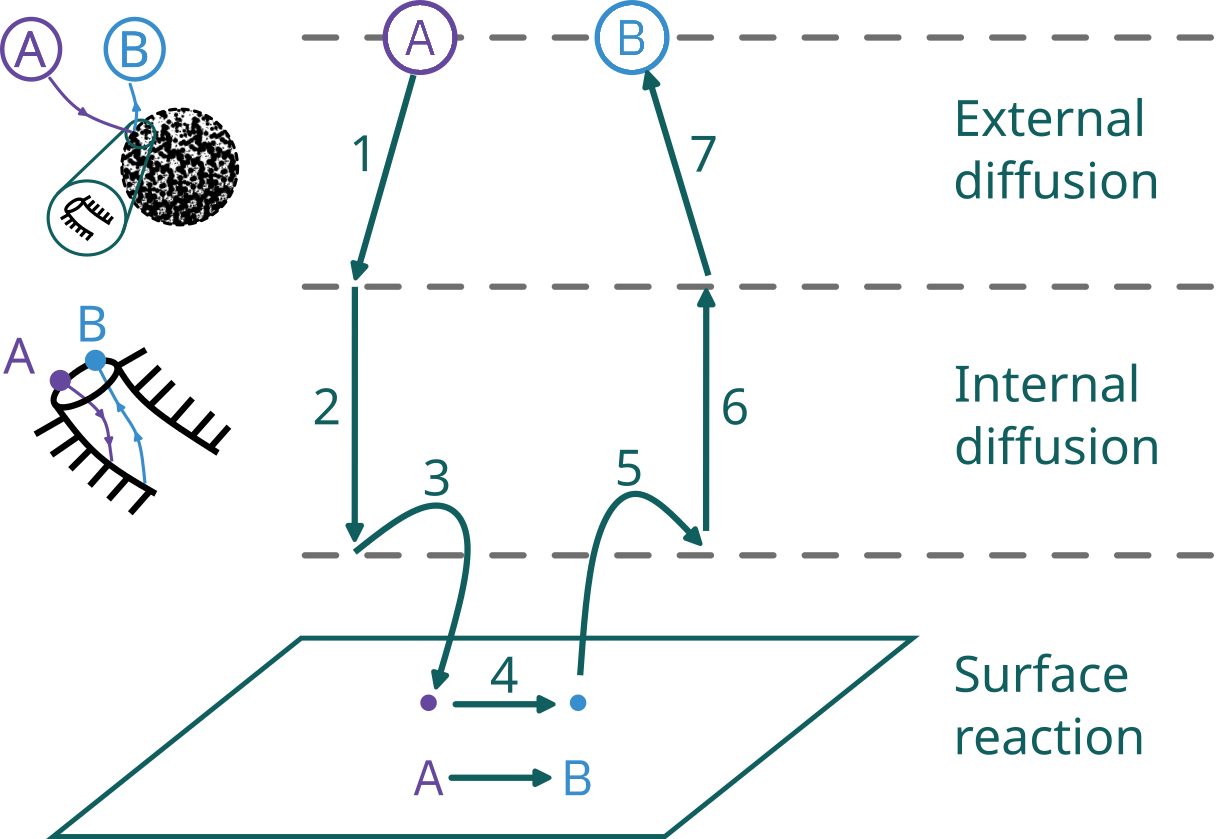
Adsorption step
Chemisorption is a necessary part of catalytic processes
Adsorption data are often presented using adsorption isotherms, which show how much gas is adsorbed by a solid at various pressures at a constant temperature.
We first propose an adsorption mechanism and derive an isotherm from it. This theoretical isotherm is then compared with experimental data.
If the theoretical isotherm aligns well with the experimental data, it suggests that the model accurately reflects the physical processes occurring within the system. Discrepancies between the predicted and actual data indicate that the model may not capture one or more crucial aspects of the physical system.
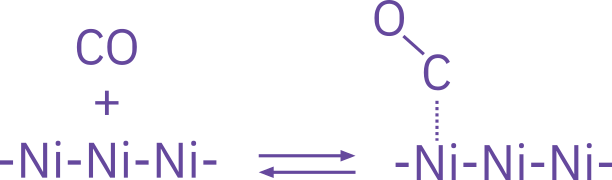
Molecular adsorption
\ce{CO + S <=> CO*S}
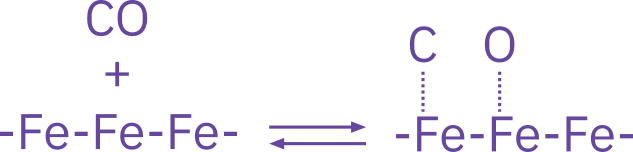
Dissociative adsorption
\ce{CO + 2S <=> C*S + O*S}
Site balance

C_t: Total number of active sites per unit mass of catalyst divided by Avogadro’s number (mol/g cat)
C_v: Number of vacant sites per unit mass of catalyst divided by Avogadro’s number
C_v is not measurable, but the total number of sites C_t can be measured
Site balance
In the absence of catalyst deactivation, assume the total number of active sites remains constant:
C_t = C_v + C_{\ce{A*S}} + C_{\ce{B*S}}
We will use the site balance equation to put C_v in terms of measurable species
Langmuir isotherm adsorption

Surface reaction step: single site
After a reactant has been adsorbed onto the surface, \ce{A*S}, is capable of reacting in a number of ways to form the reaction product
Single site mechanism
The surface reaction may be a single-site mechanism in which only the site on which the reactant is adsorbed is involved in the reaction.
An adsorbed molecule of A may isomerize (or perhaps decompose) directly on the site to which it is attached, for example, such as pentene isomerization
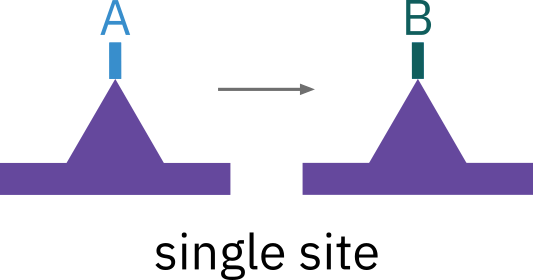
\ce{A*S <=> B*S}
- Surface reaction rate law
r_S = k_S C_{\ce{A*S}} - k_{-S} C_{\ce{B*S}}
r_S = k_S \left( C_{\ce{A*S}} -\frac{C_{\ce{B*S}}}{K_S}\right)
K_S = k_S/k_{-S}
Surface reaction step: dual site
- Mechanism 1
\ce{A*S + S <=> B*S + S}
r_S = k_S \left( C_{\ce{A*S}} C_v -\frac{C_{\ce{B*S}} C_v}{K_S}\right)
- Mechanism 2
\ce{A*S + B*S <=> C*S + D*S}
r_S = k_S \left( C_{\ce{A*S}} C_{\ce{B*S}} -\frac{C_{\ce{C*S}} C_{\ce{D*S}}}{K_S}\right)
- Mechanism 3: Two different kind of sites are involved
\ce{A*S + B*S' <=> C*S' + D*S}
r_S = k_S \left( C_{\ce{A*S}} C_{\ce{B*S'}} -\frac{C_{\ce{C*S'}} C_{\ce{D*S}}}{K_S}\right)
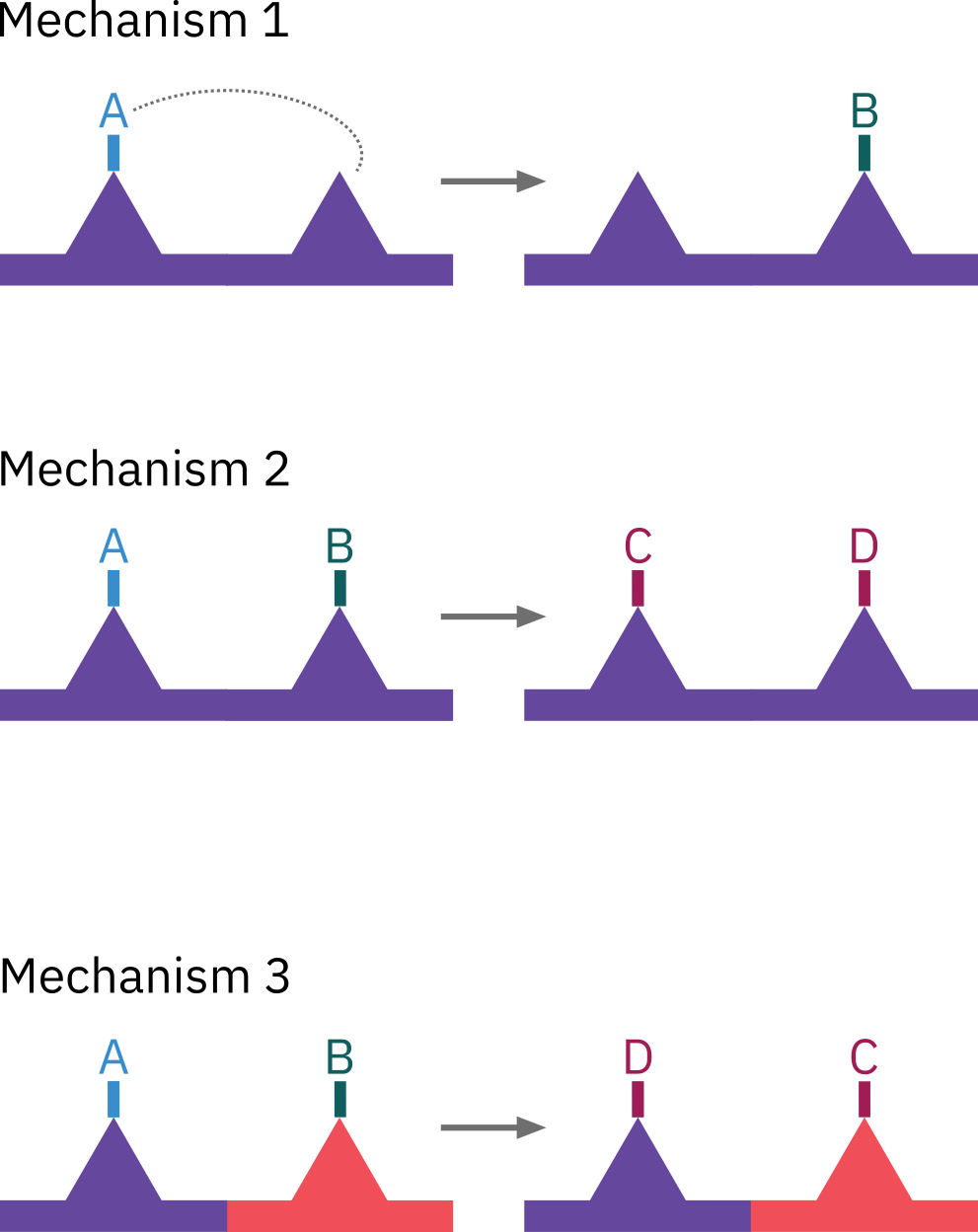
Reactions involving either single- or dual-site mechanisms, are referred to as following Langmuir-Hinshelwood kinetics.
Surface reaction step: Eley-Rideal mechanism
Reaction between adsorbed molecule and a molecule in the gas phase
Unlike the Langmuir-Hinshelwood mechanism it postulates reactions between adsorbed species and molecules directly from the gas phase.
Examples:
- The reaction of propylene and benzene
- Hydrogenation reactions: hydrogen adsorbs on the catalyst surface and reacts with an organic molecule directly striking from the gas phase.
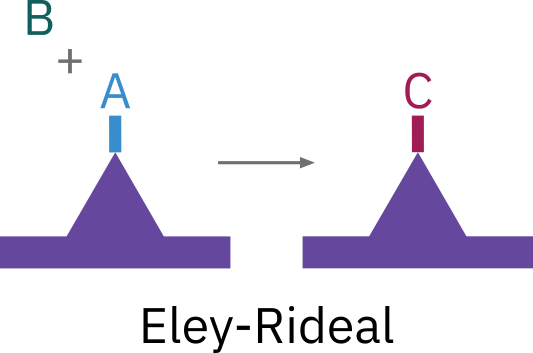
\ce{A*S + B (g) <=> C*S}
r_S = k_S \left( C_{\ce{A*S}} P_B -\frac{C_{\ce{C*S}}}{K_S}\right)
Catalyst decay/ deactivation
Solid catalyst activity changes with operation time.
- Catalyst activity usually decreases with operation time (chemical or/and physical reasons).
- sintering, fouling/ coking, poisoning
Catalyst life time is from few minutes to 10 years.
Regeneration of solid catalyst is important
Effects of catalyst decay on solid catalysed reaction
- Conversion, selectivity decreases
- Require higher reaction temperature
- Pressure drop in a fixed bed reactor increases
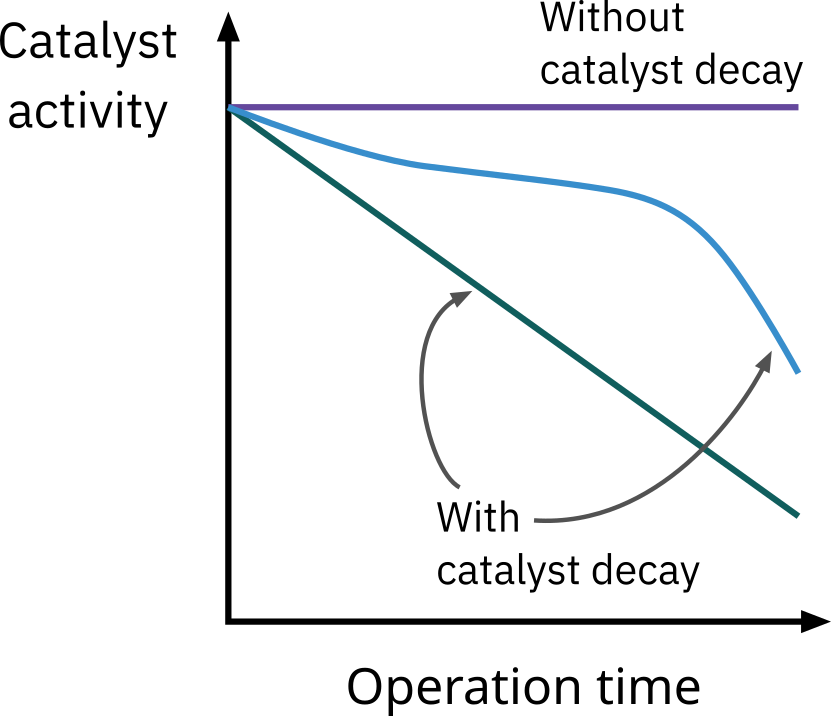
It is very rare to see catalyst activity increases with the operation time
The nickel-based catalysts used in hydroprocessing is often susceptible to sulfur poisoning. However, certain conditioning processes involving controlled exposure to sulfur compounds can rearrange the surface structure, enhancing the catalyst’s activity or selectivity over time.
Sintering
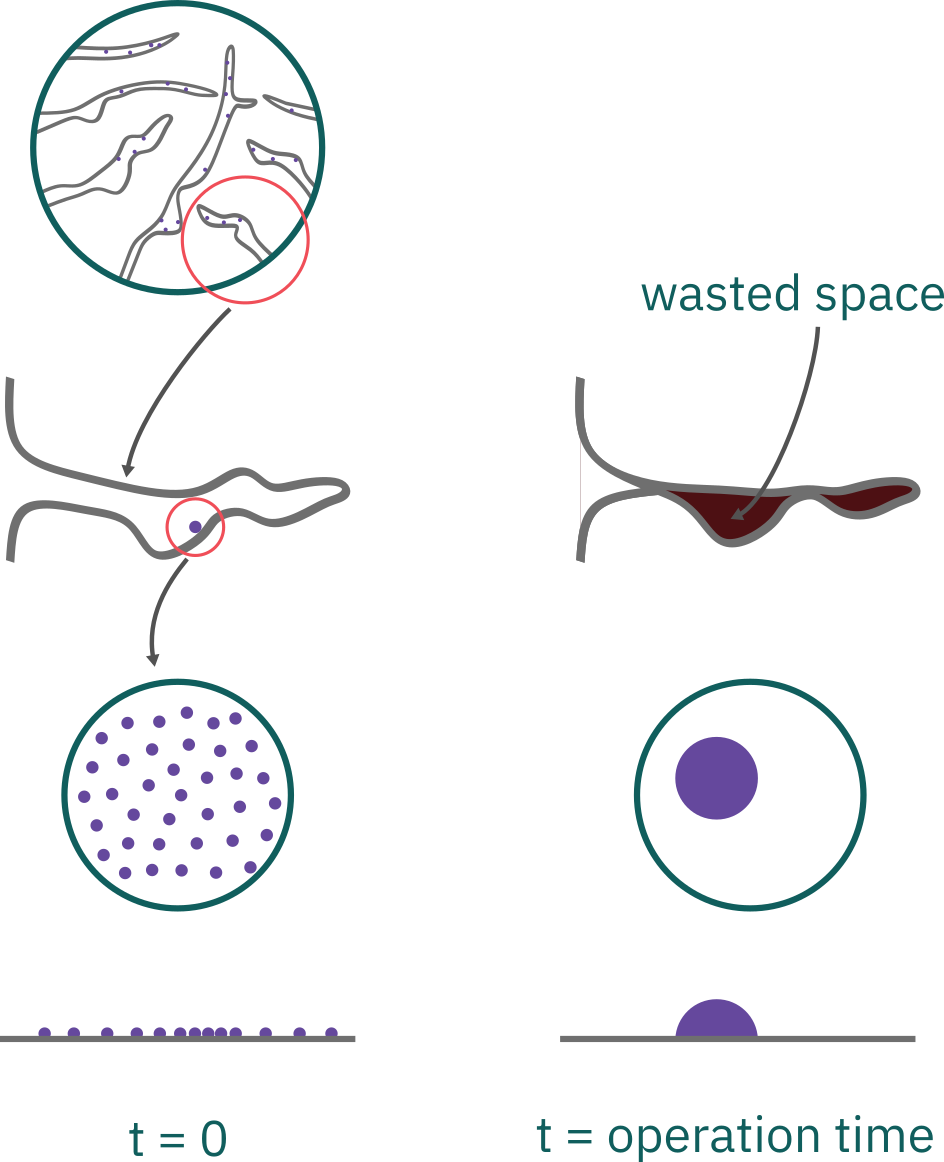
Amount of sintering is usually measured in terms of active surface area of the catalyst S_a
S_a(t) = \frac{S_{a0}}{1 + k_d t}
Loss of catalytic activity due to a loss of active surface area
- growth of the metals deposited on the support
- narrowing or closing of the pores inside the catalyst pellet
Decay law is second order with respect to the present activity
r_d = k_d a^2 = - \frac{da}{dt}
Integrating with a = 1 at time t=0 for constant k_d
a(t) = \frac{1}{1 + k_d t}
The sintering decay constant follows Arrhenius equation
k_d = k_d (T_0) \exp \left[ \frac{E_d}{R} \left( \frac{1}{T_0} - \frac{1}{T} \right) \right ]
Fouling/ Coking
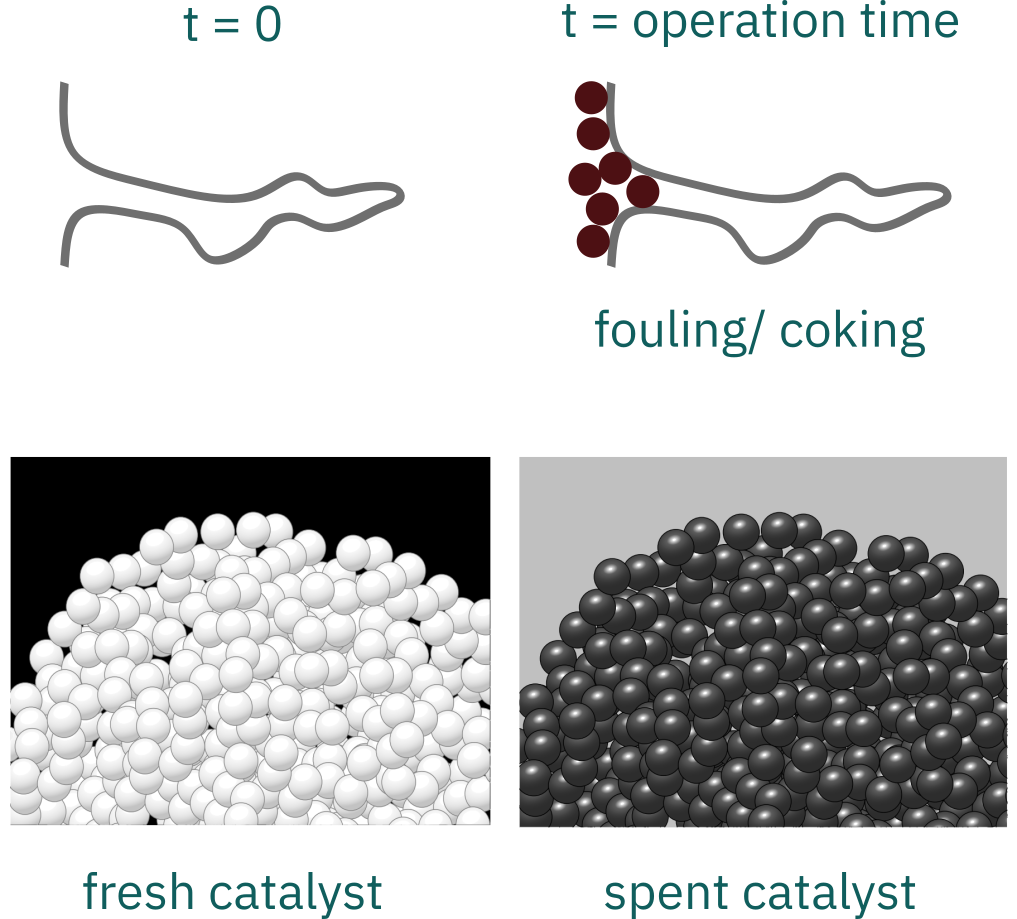
When possible, coking can be reduced by running at elevated pressures (2000–3000 kPa) and hydrogen-rich streams.
Catalysts deactivated by coking can usually be regenerated by burning off the carbon.
Results of carbonaceous (coke) material deposited on the surface of a catalyst
This mechanism is common to reactions involving hydrocarbons.
The amount of coke on the surface after a time t
C_C = A t^n
For cracking of crude oil in fixed bed
C_C = 0.52 t^{0.38}
Empirical relationships between a and C_C
\begin{aligned} a &=& \frac{1}{k_{Ck} C_C^p + 1} \\ a &=& e^{-\alpha_1 C_C} \\ a &=& \frac{1}{1 + \alpha_2 C_C} \end{aligned}
Poisoning
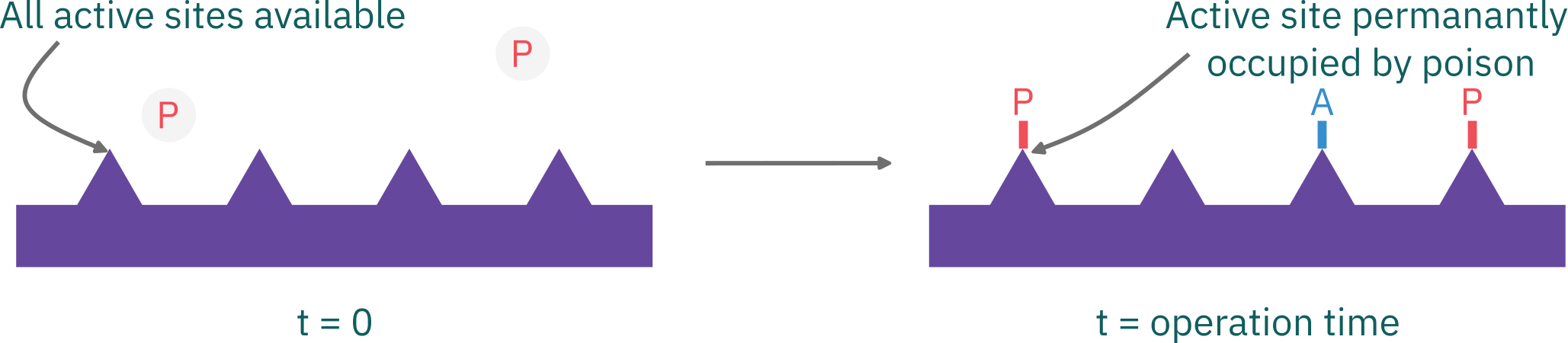
- The poisoning molecules become irreversibly chemisorbed to active sites
- Reduces the number of sites available for the main reaction.
- The poisoning molecule, P, may be a reactant and/or a product in the main reaction, or it may be an impurity in the feed stream.
\text{Main reaction:} \left\{ \begin{array}{ll} \ce{A + S <=> A*S} \\ \ce{A*S <=> B*S + C(g)} \\ \ce{B*S <=> B + S} \end{array} \right\} \quad -r'_{A} = a(t) \frac{k_{A} C_{A}}{1 + K_{A} C_{A} + K_{B} C_{B}}
\text{Poisoning reaction:} \qquad \ce{P + S -> P*S} \qquad -r'_{d} = -\frac{da}{dt} = k'_d C_p^m a^q
Catalyst regenration
