Non-isothermal reactor design
Chemical Reaction Engineering
Need for energy balance
- Consider a liquid phase PFR for a first-order exothermic reaction.
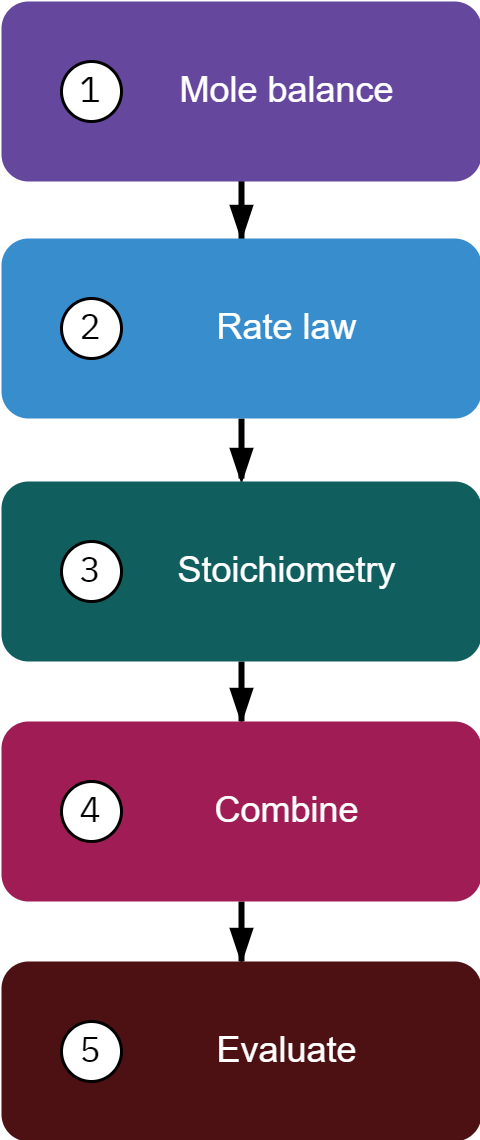
Mole balance
\frac{dX_A}{dV} = \frac{-r_A}{F_{A0}}
Rate law
-r_A = kC_A = -k_0 \exp \left[ \frac{E}{R} \left( \frac{1}{T_1} - \frac{1}{T} \right)\right] C_A
Stoichiometry
C_A = C_{A0}(1-X_A)
Combine: Put F_{A0} in terms of C_{A0}
\frac{dX}{dV} = -k_0 \exp \left[ \frac{E}{R} \left( \frac{1}{T_1} - \frac{1}{T} \right)\right] \frac{(1 - X_A)}{\upsilon_0}
Evaluate
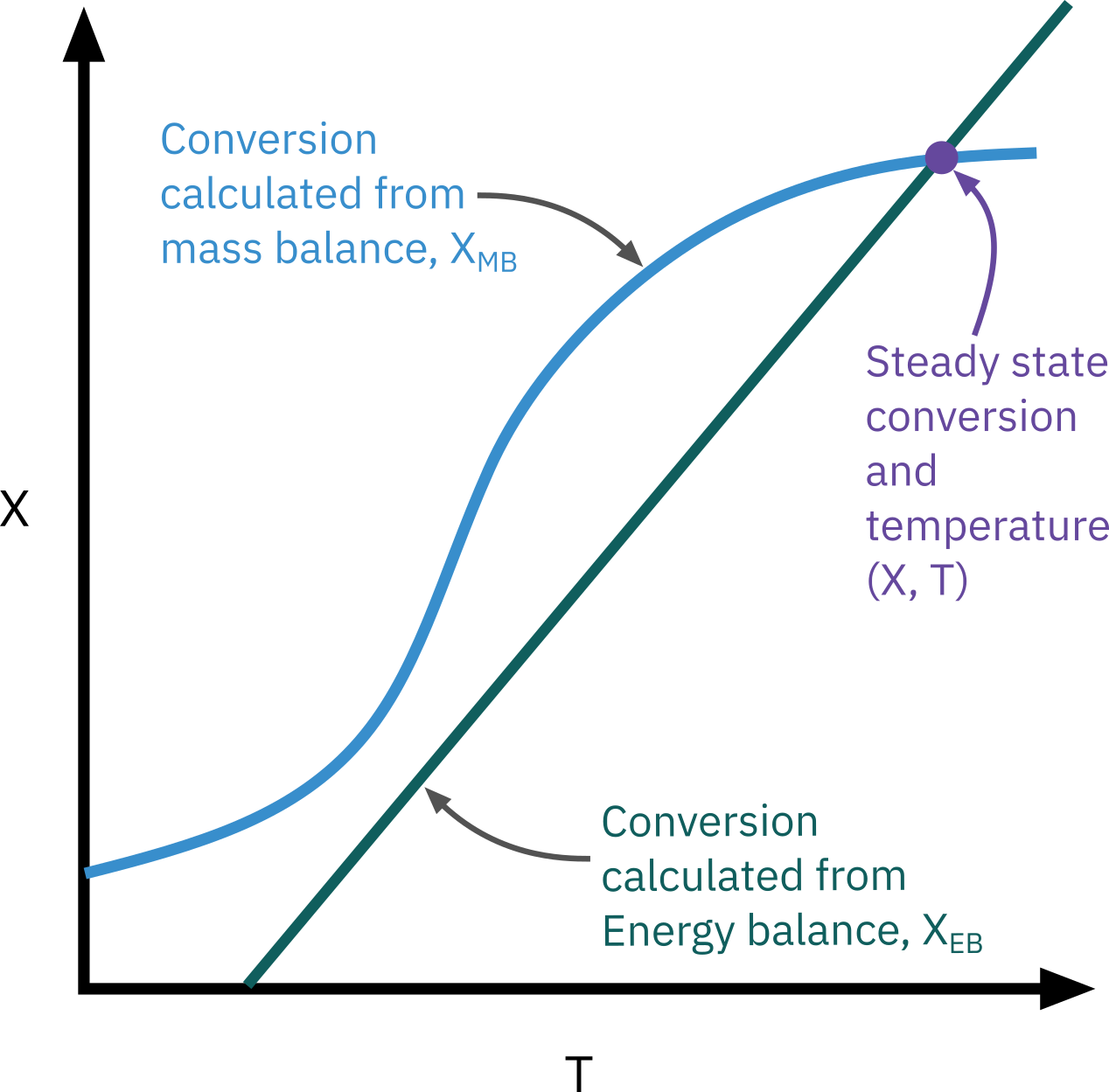
We cannot solve this equation because both X, and T vary with V and we don’t have X either as a function of V or T. We need one more equation
\Rightarrow The energy balance
Thermodynamics in a closed system
First law of thermodynamics
System
A system is any bounded portion of the universe, moving or stationary, which is chosen for the application of the various thermodynamic equations.
Closed system: no mass crosses boundaries
\begin{aligned} \text{Change in} & & \text{Heat flow} & & \text{Work done by} \\ \text{total energy} &\;\; = & \text{to} \qquad &\;\; - & \text{by the system} \\ \text{of the system} & & \text{the system} & & \text{on the surroundings} \end{aligned}
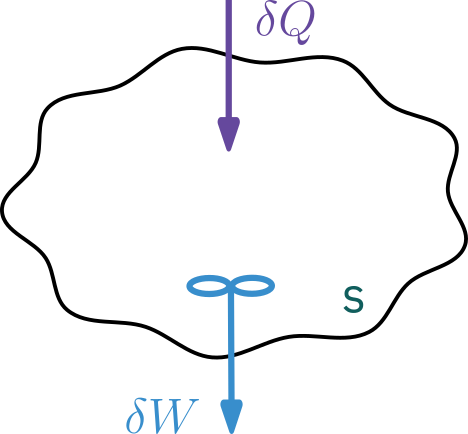
d \hat{E} = \delta Q - \delta W
- \hat{E}: Change in total energy of the system
- \delta Q: Heat flow to the system
- \delta : Work done by system on the surroundings
Thermodynamics in an open system
- Open system: continuous flow system, mass crosses the system’s boundaries
- Mass flow can add or remove energy
- System volume is well mixed
- N species each entering and leaving at molar flow rate of F_i and has internal energy of E_i
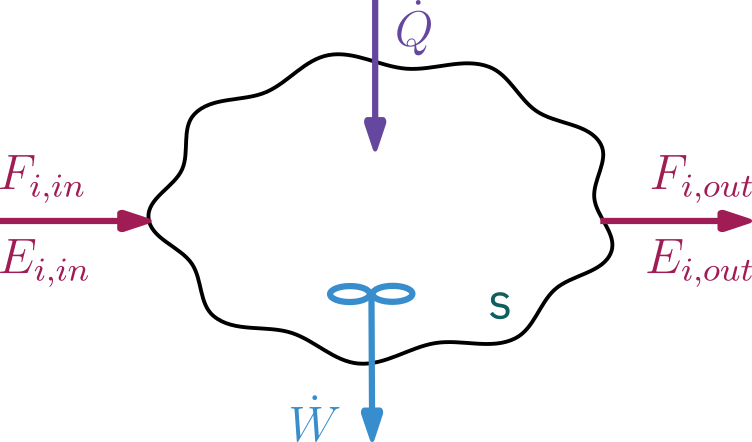
\begin{aligned} \text{Rate of accumulation} & & & & \text{Work done} & & \text{Energy added} & & \text{Energy leaving} \\ \text{of energy} &\;\; = & \text{Heat in} &\;\; - & \text{by} \qquad &\;\; + & \text{to the system} &\;\; - & \text{the system by} \\ \text{in the system} & & & & \text{the system} & & \text{by mass flow in} & & \text{mass flow out} \end{aligned}
\frac{d \hat{E}_{sys}}{dt} = \dot Q - \dot W + \left. \sum_{i=1}^{N} E_i F_i \right|_{in} - \left. \sum_{i=1}^{N} E_i F_i \right|_{out}
Steady state energy balance
Constant heat capacity, adiabatic system
X_{EB} = \frac { \sum \Theta_i C_{P_i} \left[ T_{i} - T_{0}\right] } {- \Delta H_{Rx}^\circ (T_R) + \Delta C_{P}(T - T_R) }
- In many instances, the \Delta C_{P}(T - T_R) term in the denominator is negligible with respect to the \Delta H_{Rx}^\circ (T_R) term,
T = T_0 + \frac{ \left( -\Delta H^\circ_{Rx} \right) X_{EB}}{\sum \Theta_i C_{Pi}}

This equation applies to CSTR, PFR, PBR, and Batch reactors. It gives us the explicit relationship between X and T needed to be used in conjunction with the mole balance to solve a large variety of chemical reaction engineering problems.
Adiabatic equilibrium conversion
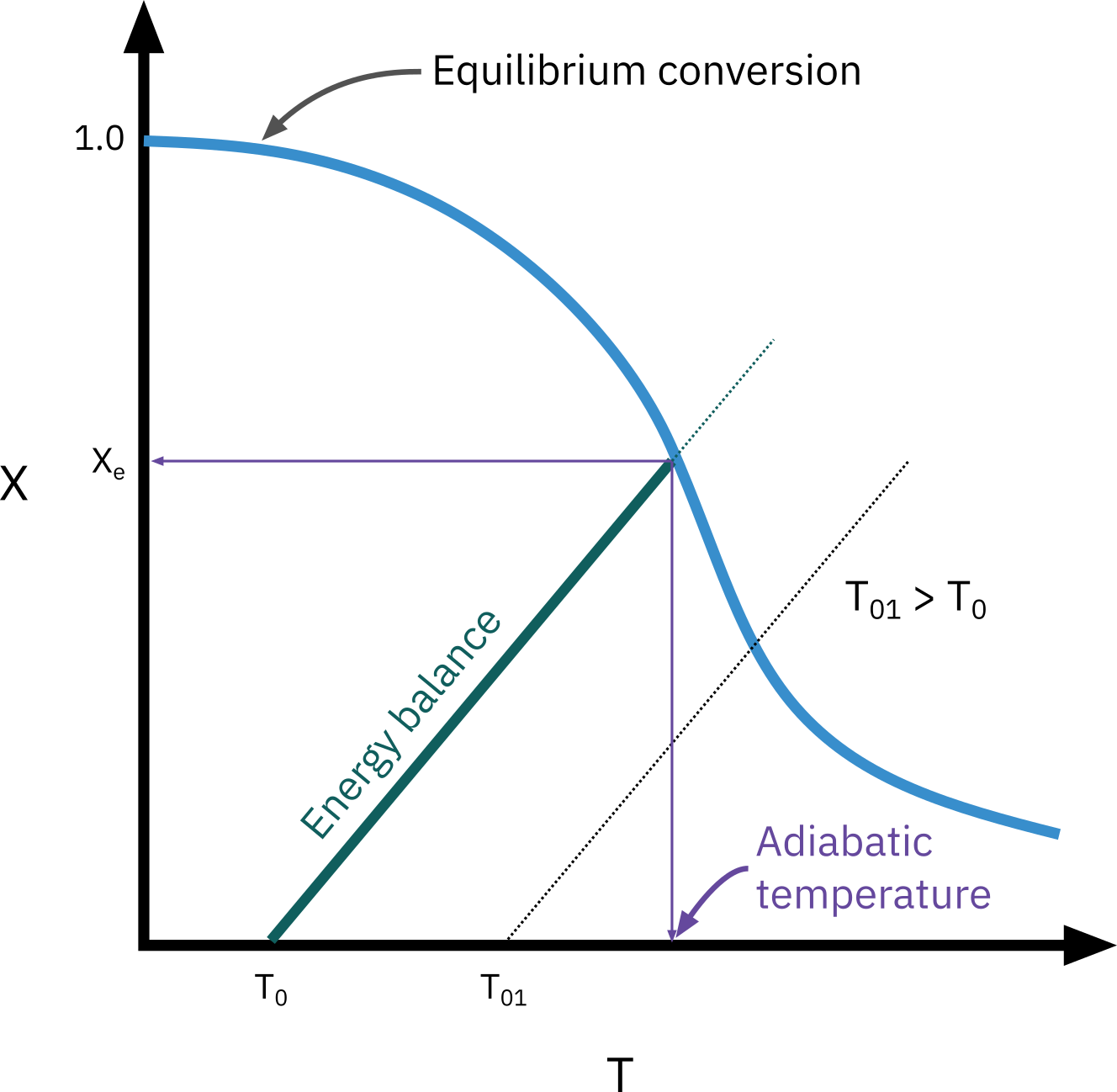
Exothermic reactions
Endothermic reactions
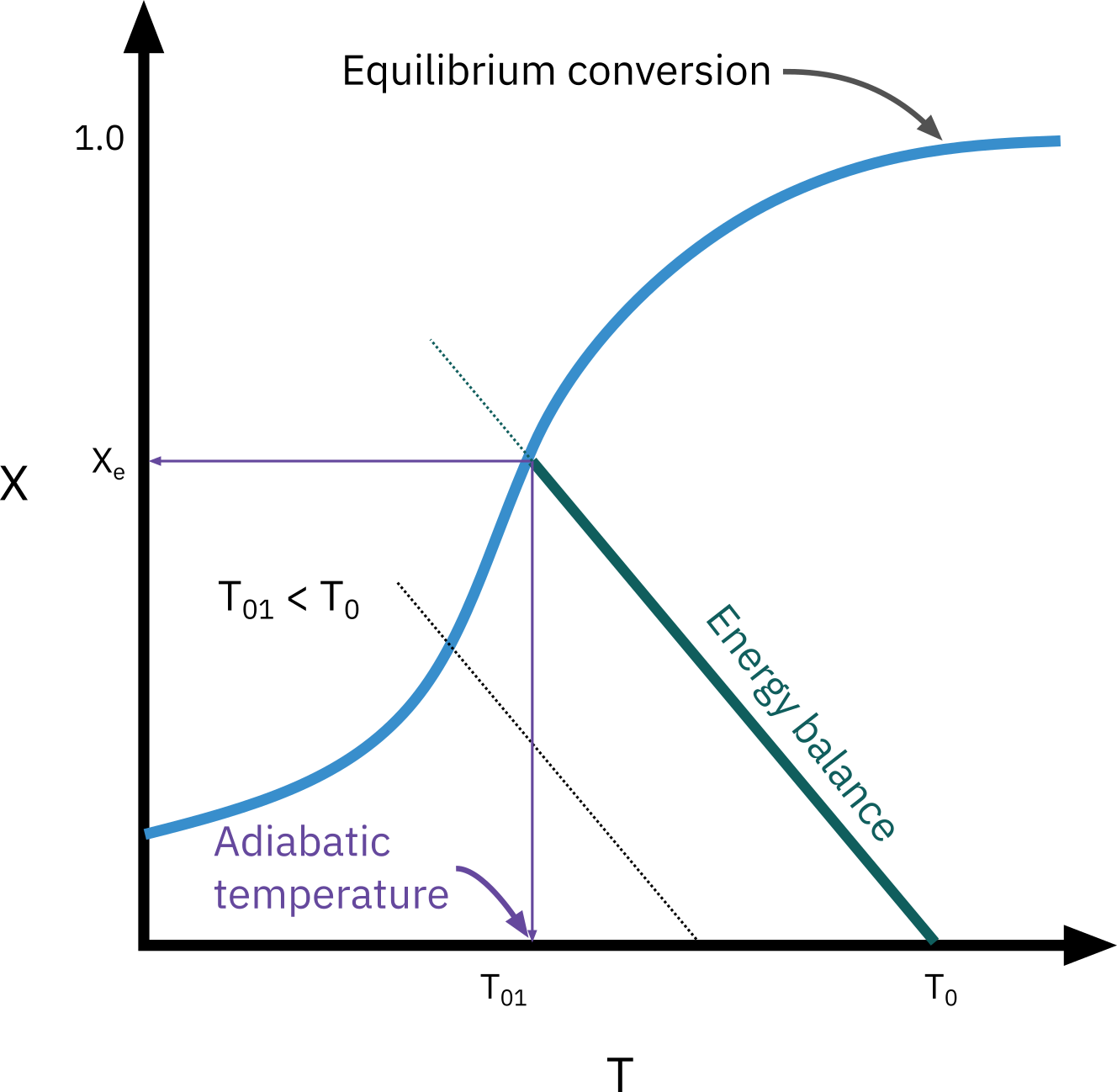
The highest conversion that can be achieved in reversible reactions is the equilibrium conversion. For endothermic reactions, the equilibrium conversion increases with increasing temperature up to a maximum of 1.0. For exothermic reactions, the equilibrium conversion decreases with increasing temperature.
Reactor staging
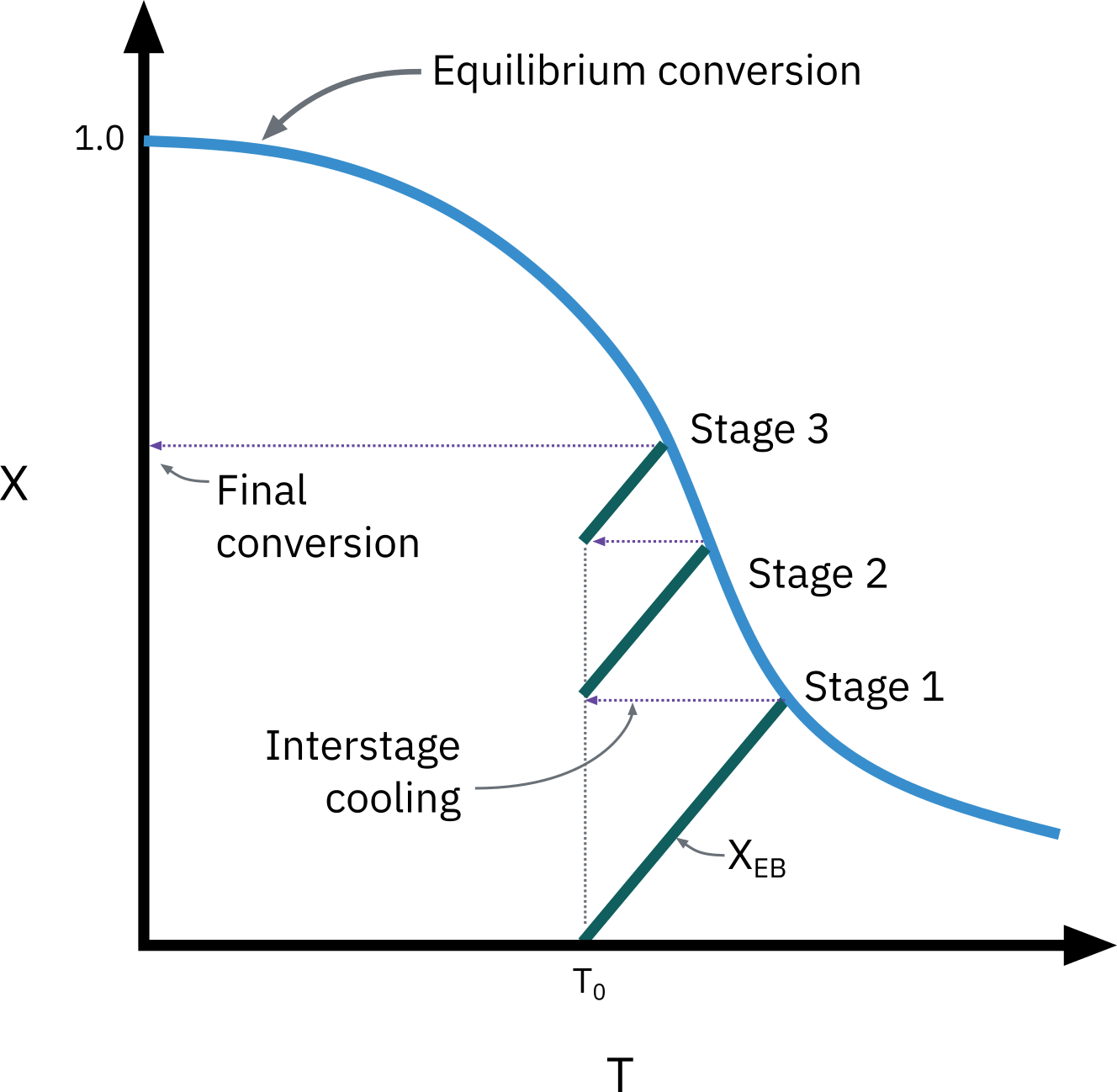
Exothermic reactions
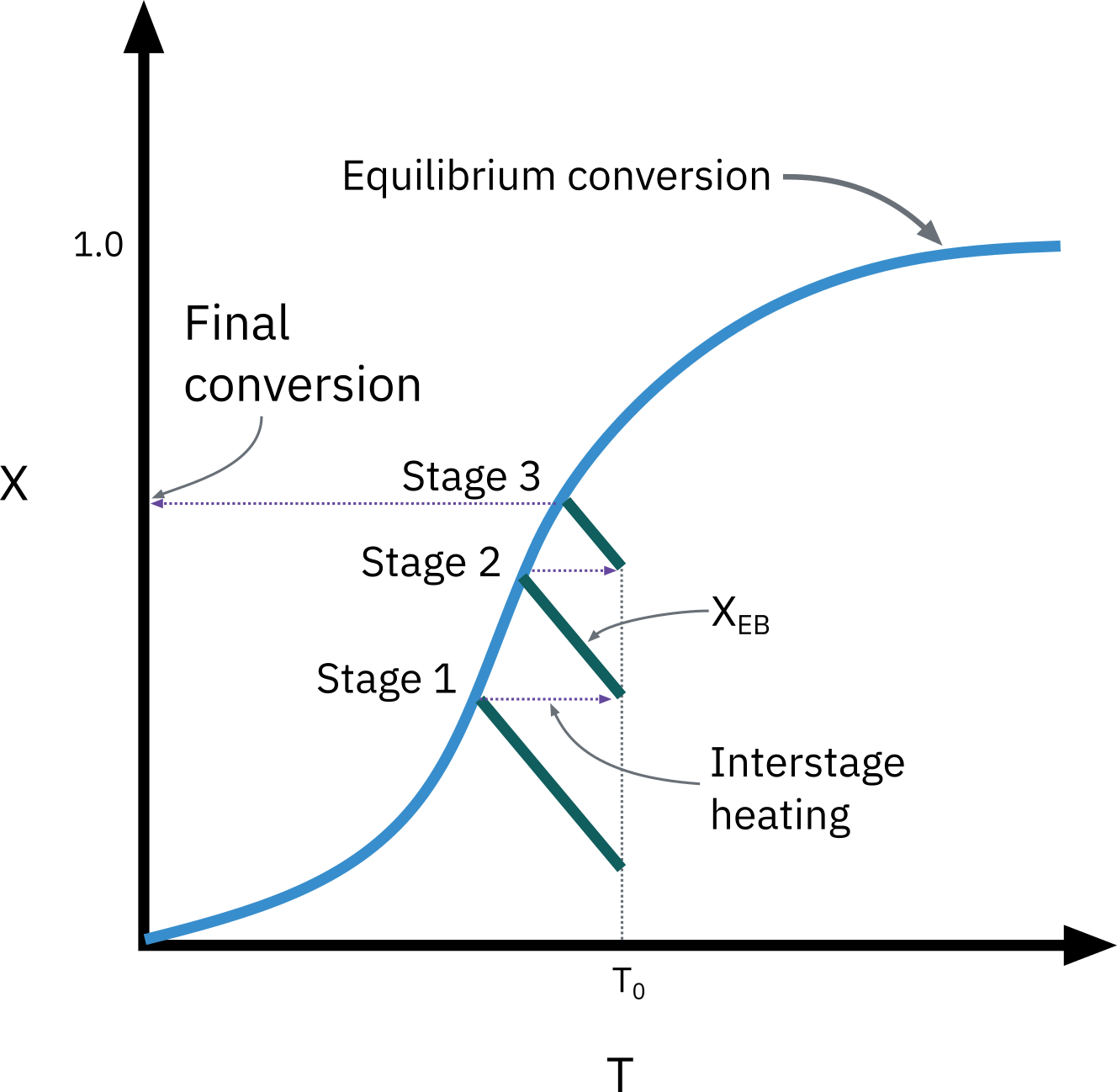
Endothermic reactions

Optimum feed temperature
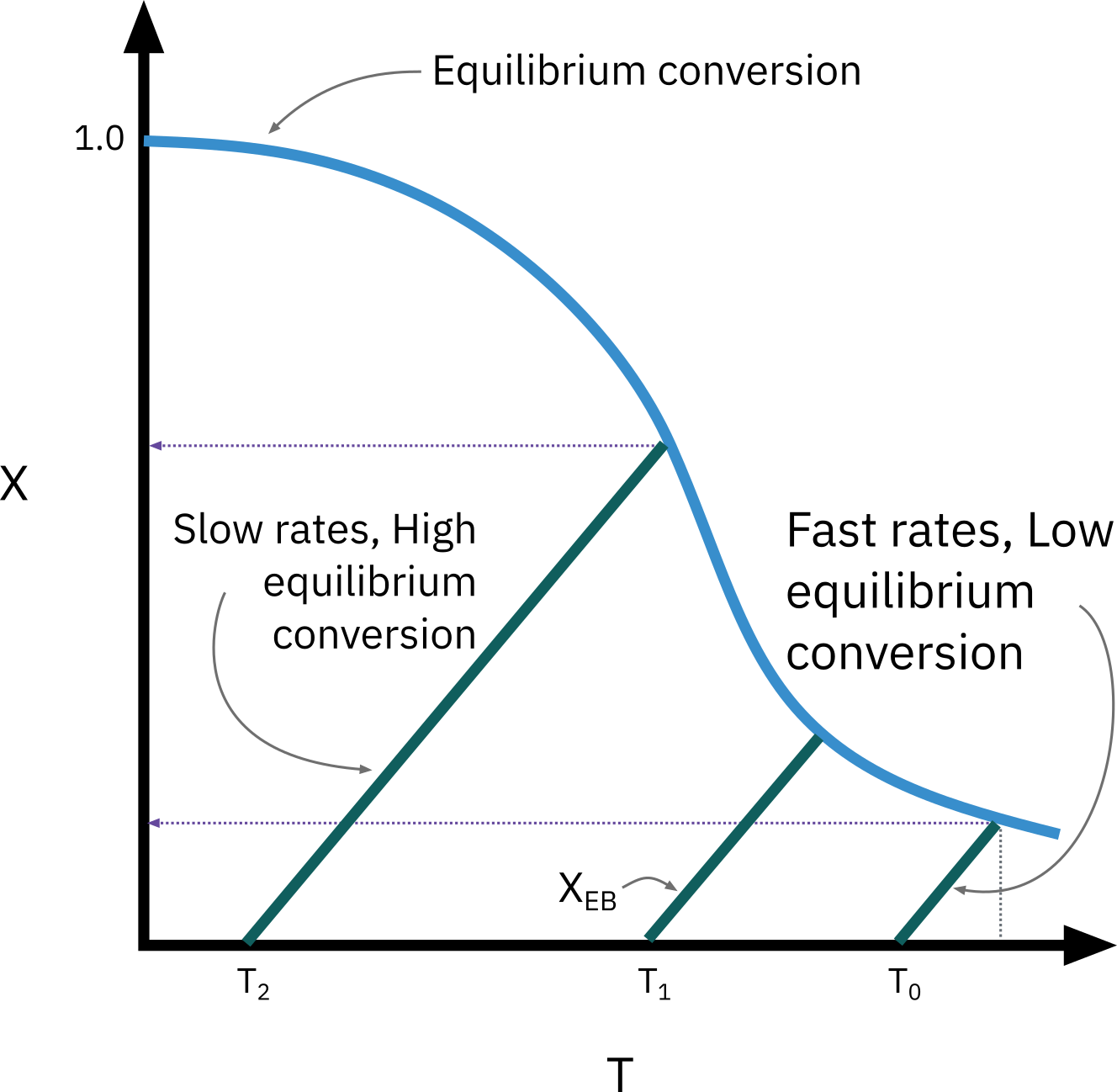
Optimum feed temperature

Optimum feed temperature

Optimum feed temperature

Optimum feed temperature

Optimum feed temperature

Steady-state tubular reactor with heat exchange
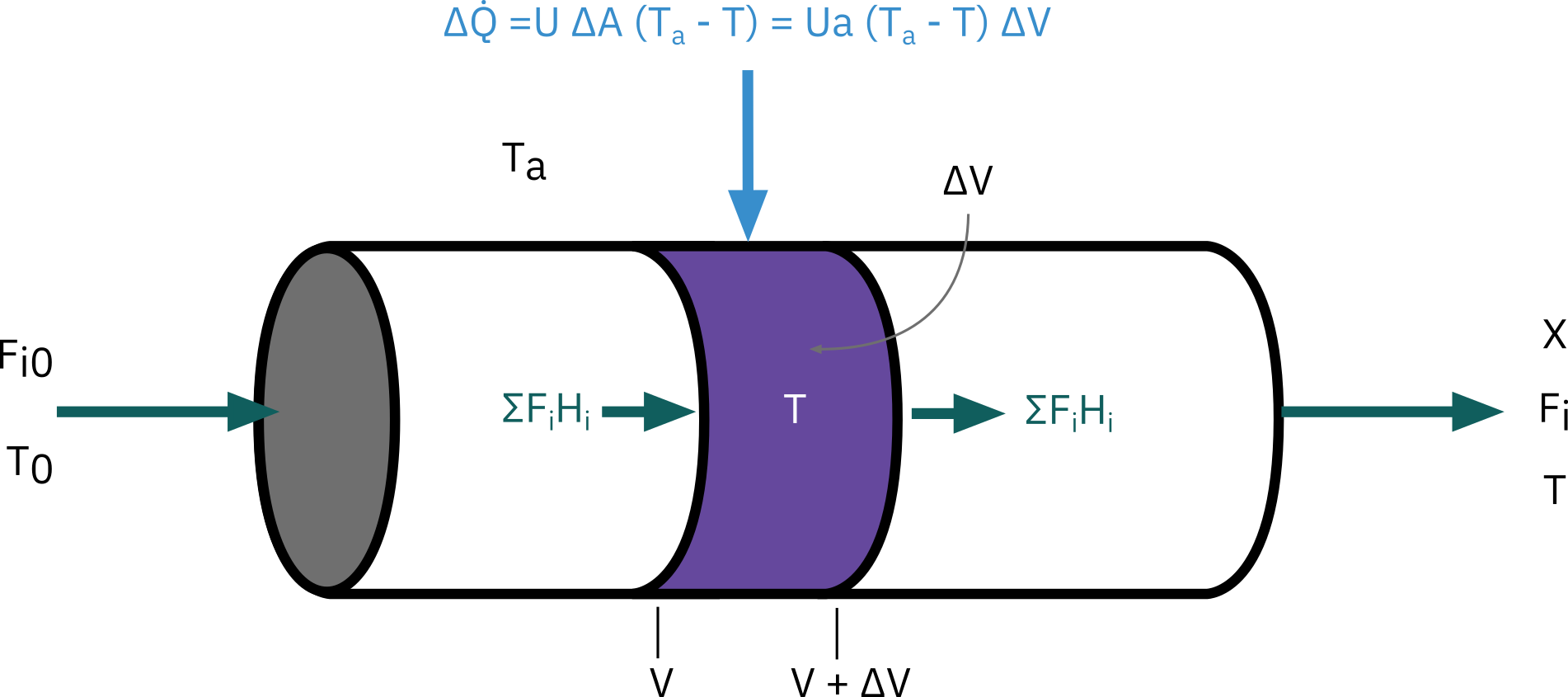
Energy balance over volume \Delta V
\Delta \dot Q + \sum F_i H_i |_V - \sum F_i H_i |_{V + \Delta V} = 0
Heat flow into the reactor \Delta \dot Q
\Delta \dot Q = U \Delta A (T_a - T) = Ua \Delta V (T_a - T)
For tubular reactor a = 4/D
- Dividing by \Delta V and taking limit as \Delta V \rightarrow 0
Ua (T_a - T) - \frac {d\sum F_i H_i}{dV} = 0
Balance on the heat-transfer fluid
The heat-transfer fluid will be a coolant for exothermic reactions and a heating medium for endothermic reactions.
If the flow rate of the heat-transfer fluid is sufficiently high with respect to the heat released (or absorbed) by the reacting mixture, then the heat-transfer fluid temperature will be virtually constant along the reactor. For all other cases, we need to write energy balance for the heat transfer fluid.
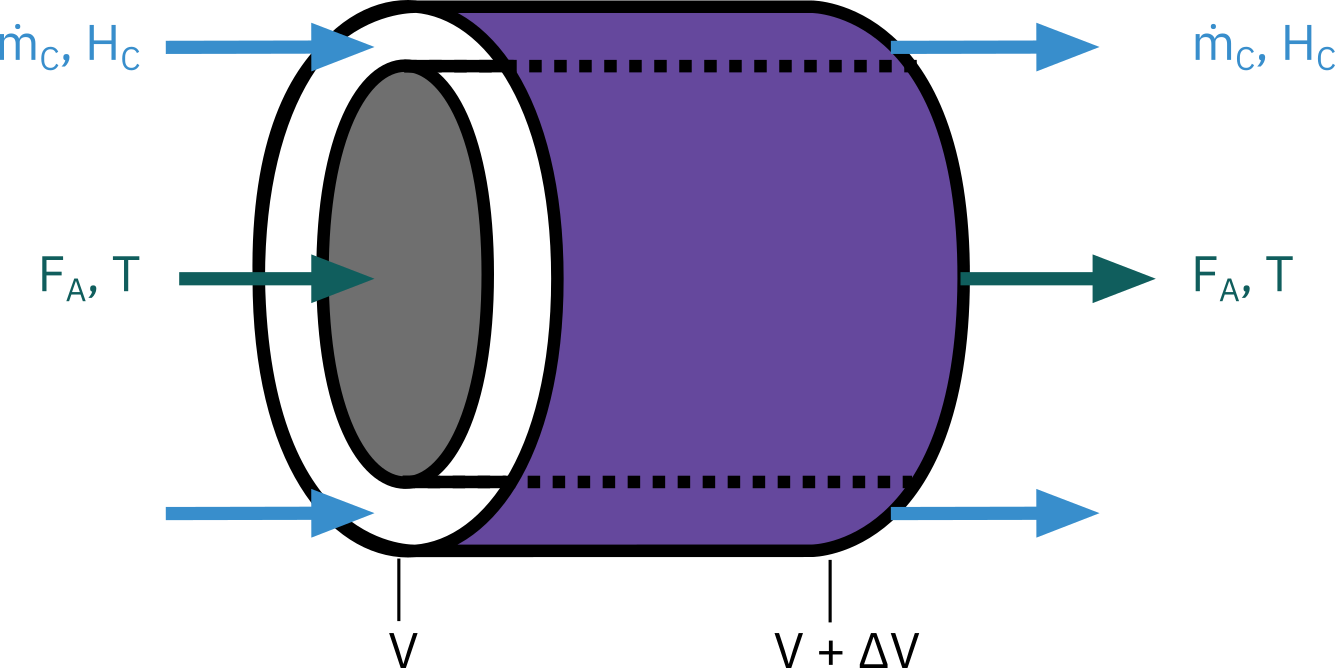
\begin{aligned} \text{Rate of energy} & & \text{Rate of energy} & & \text{Rate of heat added} & & \\ \text{in at } V & \;\; - & \text{out at } V + \Delta V & \;\; + & \text{by conduction through} & \;\; = & 0 \\ & & & & \text{the inner wall} & & \end{aligned}
\dot m_C H_C |_V - \dot m_C H_C |_{ V + \Delta V } + \underbrace{U a (T - T_a) \Delta V}_{\color{RoyalBlue}{\text{Cocurrent flow}}} = 0
\frac{dT_a}{dV} = \frac{U a (T - T_a)} { \dot m_C C_{P_C} }
The \dot Q Term in the CSTR
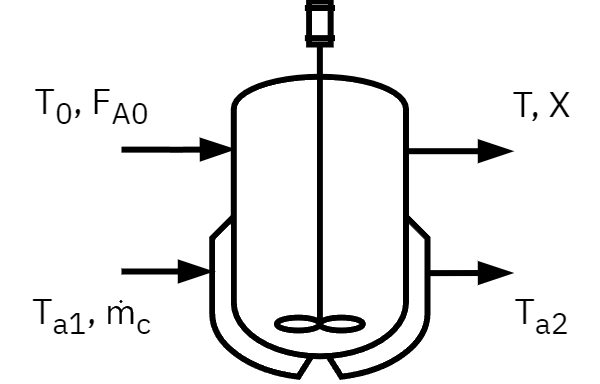
Rate of energy in by flow:
\dot m_c C_{P,c} (T_{a1} - T_R)
Rate of energy out by flow:
\dot m_c C_{P,c} (T_{a2} - T_R)
Rate of heat transfer from exchanger to reactor:
\frac{UA(T_{a1} - T_{a2})}{\ln{\left[\frac{(T - T_{a1})} {(T - T_{a2})}\right]}}
- An energy balance on the heat-exchanger fluid entering and leaving the exchanger
\begin{aligned} \text{Rate of} & & \text{Rate of} & & \text{Rate of} & & \\ \text{energy} &\;\; - & \text{energy} &\;\; - & \text{heat transfer} &\;\; = & \text{0} \\ \text{in} & & \text{out} & & \text{from exchanger} & & \\ \text{by flow} & & \text{by flow} & & \text{to reactor} & & \end{aligned}
\dot{Q} = \dot{m}_c C_{P,c} (T_{a1} - T_{a2}) = \frac{UA (T_{a1} - T_{a2})} {\ln\left(\frac{T - T_{a1}}{T - T_{a2}}\right)}
T_{a2} = T - (T - T_{a1}) \exp \left( \frac{-UA}{\dot{m}_c C_{P,c}} \right)
\dot{Q} = \dot{m}_c C_{P,c} \left( T_{a1} - T \right) \left[ 1 - \exp \left( \frac{-UA}{\dot{m}_c C_{P,c}} \right) \right]
For very large \dot m_c
\dot{Q} = U A (T_a - T)
Conversion in a non-adiabatic CSTR